
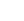

CASES OF THE WEEK – “Alport’s syndrome: a rare cause of Nephrotic Syndrome” by Dr Aditya Bhabhe, Consultant Nephrology
Alport’s syndrome: a rare cause of Nephrotic Syndrome.
19 year old female patient presented with complains of headache since 2 weeks and high blood pressure readings. She had no significant past medical history, except for sensori-neural deafness which was treated with hearing aid. There was no family history of kidney disease.
On examination the patient’s blood pressure was elevated: 140/94 and she had minimal pedal edema.
Systemic examination was within normal limits. Her labs showed elevated serum creatinine: 1.4 mg/dL ; urinalysis showed proteinuria. 24 hour urine protein excretion was very high: 4277 mg (Normal < 30 mg/day). She also had low serum albumin and hyperlipidemia. Her CBC, Liver function tests and electrolytes were normal. USG showed normal sized kidneys.
A provisional diagnosis of Nephrotic syndrome was reached. Auto immune work up including ANA, cANCA, pANCA and C3 and C4 was normal. The patient underwent a CT guided kidney biopsy and the samples were analyzed by light microscopy, immunofluorescence and electron microscopy. Common causes of Nephrotic syndrome in this age group are Minimal change disease, Membranous Glomerulonephritis , FSGS and MPGN.
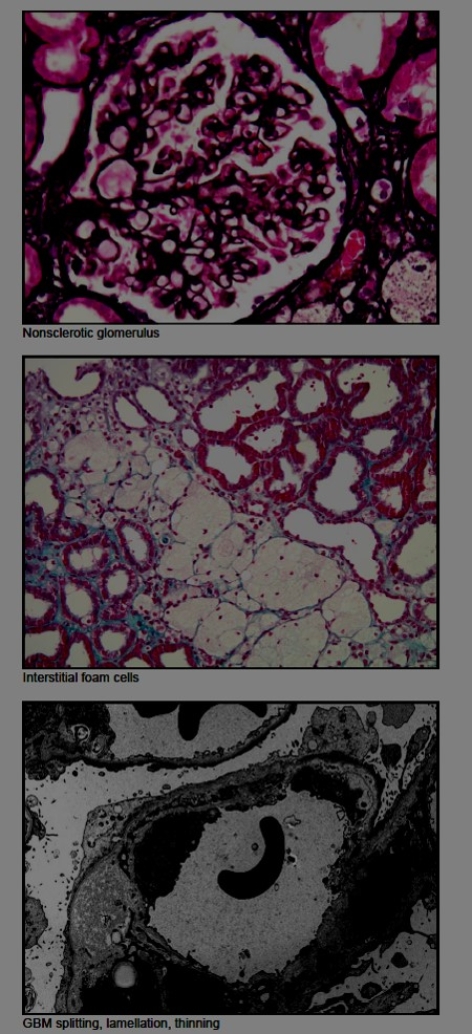
However, to our surprise, the kidney biopsy showed: Sclerosing glomerulopathy with abnormalities in glomerular basement membrane architecture, highly suggestive of hereditary nephritis/Alport nephropathy.
Patient and her family were counseled about the nature of the disease and she was started on ACE inhibitors. Her proteinuria and blood pressure are presently improving. She remains at risk for progression to advanced CKD.
Discussion:
Alport syndrome is a rare genetic disorder characterized by progressive kidney disease and abnormalities of the inner ear and the eye. Alport syndrome accounts for only 3% of children with chronic kidney disease and 0.2% of adults with end-stage renal disease. It is inherited as Autosomal recessive or X Linked dominant or recessive disorder. Renal disease can be conclusively diagnosed only by Electron microscopic examination of kidney biopsy sample.
Treatment is symptomatic including use of RAAS blockers. Newer gene therapies are under evaluation. Pateints can progress to ESRD requiring dialysis or kidney transplantation.
In summary, it is imperative that all adults with nephrotic syndrome undergo a kidney biopsy and have a detailed analysis of the biopsy tissue including electron microscopy, so as to not miss such rare causes of kidney disease.